Classification: The First and Most Critical Step in the Regulatory Process
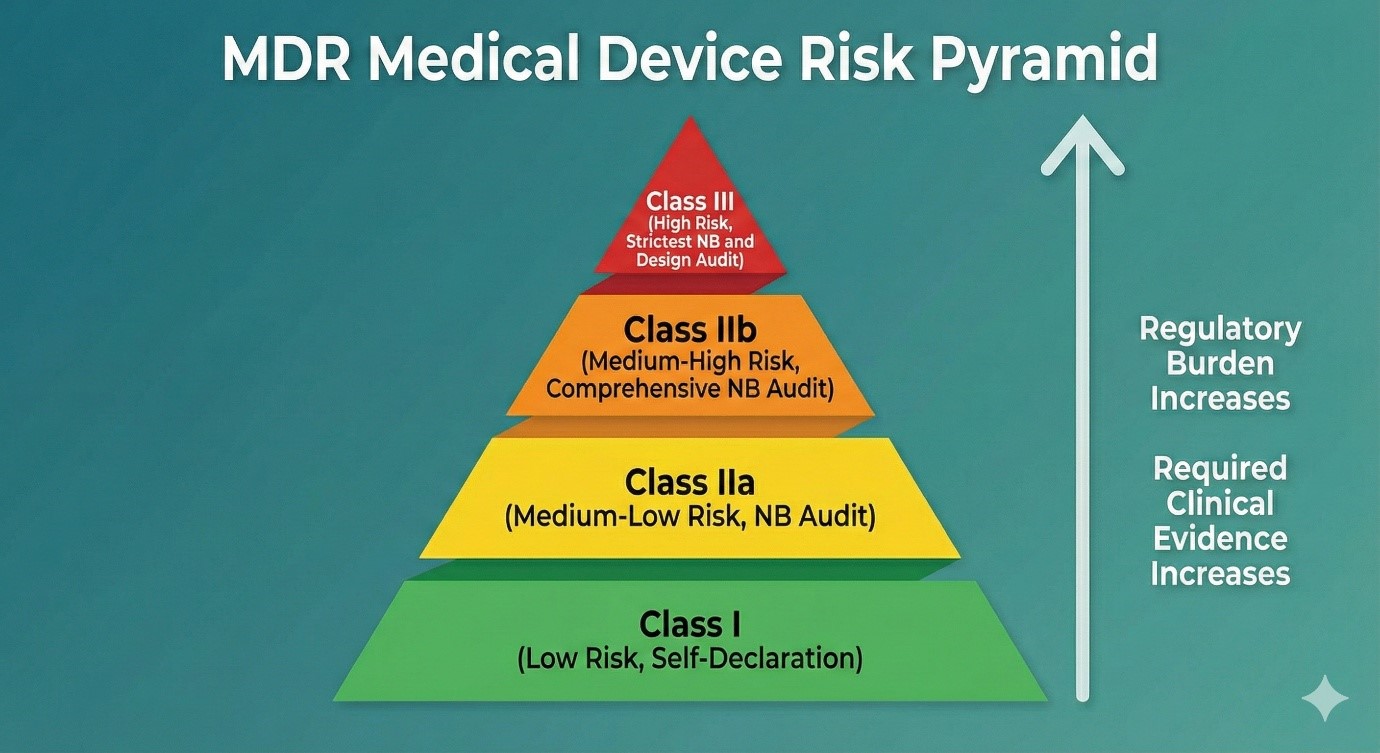
In the previous sections, we examined the definition of a medical device and the new regulatory framework introduced by the MDR/IVDR. The first and most critical step in determining a device’s regulatory roadmap is the accurate determination of its risk class. The European Union Medical Device Regulation (MDR) divides devices into four main classes based on their potential risk: Class I, Class IIa, Class IIb, and Class III.
This risk-based approach directly determines the complexity of the device’s approval process, the amount of clinical evidence required, and the scope of Notified Body (NB) oversight. A high-risk device is naturally subjected to a much more rigorous review than a low-risk device.
Fundamental Principles of Risk Classification
The 22 rules contained in Annex VIII of the MDR establish the fundamental criteria used for classifying devices. These rules take into account the following characteristics of the device:
- Invasiveness (Entry into the Body): Whether the device enters the body and by what route (surgical, natural orifice, etc.).
- Duration of Use: How long the device remains in the body (transient, short-term, long-term).
- Local Effect: The device’s interaction with critical areas of the body such as the central circulatory system, central nervous system, or heart.
- Energy Source: Whether the device is active and the potential risk of its energy source.

The Four Risk Classes and Their Regulatory Implications
Class | Risk Level | Example Devices | Approval Pathway (CE Mark) | Notified Body (NB) Oversight |
Class I | Low Risk | Spectacle frames, wound dressings, reusable surgical instruments. | Manufacturer’s Self-Declaration (Excluding sterile or measuring function devices). | Not Required (Only relevant parts are audited for sterile or measuring function devices). |
Class IIa | Medium-Low Risk | Contact lenses, dental filling materials, ultrasound devices. | NB Audit (Sampling of QMS and Technical File). | Mandatory (Lighter audit). |
Class IIb | Medium-High Risk | Blood transfusion sets, certain ventilators, bone cements. | NB Audit (More comprehensive QMS and Technical File review). | Mandatory (Comprehensive audit). |
Class III | High Risk | Heart valves, permanent implants, artificial organs, devices containing biological material. | NB Audit (Strictest QMS and Technical File review, design examination). | Mandatory (Highest level of oversight). |
Impact of Classification on Clinical Investigations
The risk class of the device directly determines the quantity and type of clinical evidence required.
- Class I Devices: They generally do not require extensive clinical research. Existing literature data, equivalence, safety reports, and technical documentation are considered sufficient.
Class IIa- Medium-Risk Medical Devices: These are medium-risk devices that are inserted into the body for a short period of time or used for measurement/support purposes. Performance studies and safety data are required when necessary. Observational or small-scale clinical studies may be conducted.
Class IIB- High-Medium Risk Medical Devices: These are high-medium risk devices that remain inside the body for a longer period of time or have a therapeutic effect. Clinical research is required. Performance tests + prospective clinical studies are conducted. TİTCK approval is mandatory.


Class III Devices: Because they are in the highest risk group, new clinical data obtained from the manufacturer’s own device (i.e., clinical studies) are often mandatory. Equivalence claims are much more difficult and limited. These are the highest-risk devices that are permanent and support vital functions. Comprehensive clinical studies are required. Randomized controlled trials, long-term safety data, and multi-center studies are required. Approval must be obtained from the Turkish Medicines and Medical Devices Agency (TİTCK).
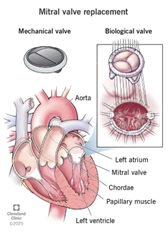
This situation directly affects the speed and cost of clinical investigation processes, especially for Class III device manufacturers.
Conclusion
Medical device classification forms the foundation of the regulatory process. Correct classification prevents manufacturers from incurring unnecessary costs and time delays on the path to the CE mark. In the next section, we will detail the process that follows this classification and is key to MDR compliance: The Path to the CE Mark and Technical Documentation.
References
[1] Regulation (EU) 2017/745 of the European Parliament and of the Council of 5 April 2017 on medical devices.
[2] MDCG 2021-24 Guidance on classification of medical devices.European Commission.