
Regulatory Transformation: The Meaning of the Transition from MDD to MDR
The European Union has introduced two new, much more comprehensive and stringent regulations to enhance the safety and performance of medical devices, replacing the former Medical Device Directives (MDD): the Medical Device Regulation (MDR) (EU) 2017/745 [1] and the In Vitro Diagnostic Medical Device Regulation (IVDR) (EU) 2017/746 [2].
The MDR became fully applicable on May 26, 2021, and the IVDR on May 26, 2022. This transition is referred to as a “regulatory revolution” in the industry and signifies fundamental changes for all stakeholders, from manufacturers to clinical researchers.
- Medical Device Regulation (MDR) – EU 2017/745
The primary goal of the MDR is to maximize patient safety and ensure transparency and traceability throughout the entire lifecycle of devices. The most significant innovations introduced by the MDR compared to the previous MDD are:
- Strengthening of Clinical Evidence
The MDR has significantly increased the quantity and quality of clinical evidence that devices must present before and after being placed on the market. Manufacturers are now required to submit more Clinical Evaluation Reports (CER) and, where necessary, Clinical Investigation data to prove that their devices are safe and performant for their intended purpose [3].
- Increased Role of Notified Bodies (NB)
The MDR has expanded the audit powers and responsibilities of Notified Bodies. NBs are required to scrutinize manufacturers’ Technical Documentation and Quality Management Systems (QMS) more rigorously and in detail. Furthermore, a portion of Class I devices may now also be subject to NB oversight.
- Traceability and Transparency (UDI and EUDAMED)
- UDI (Unique Device Identification): A requirement has been introduced to assign a unique identifier to every device, enabling tracking from production to the patient.
- EUDAMED: The European Database on Medical Devices is a centralized system where data on devices, manufacturers, clinical investigations, and post-market surveillance are recorded transparently and made publicly available.
- In Vitro Diagnostic Medical Device Regulation (IVDR) – EU 2017/746
The IVDR regulates devices used to test samples taken from the human body, such as blood, tissue, and urine (In Vitro Diagnostic Devices – IVDs). The IVDR has brought about a similar transformation for IVDs as the MDR did for medical devices.
- Risk-Based Classification
While most IVDs were placed on the market via self-declaration under the previous Directive, the IVDR introduced a new risk-based classification system (A to D). This has significantly increased the proportion of high-risk IVDs (e.g., blood screening tests) that require Notified Body oversight.
- Strengthening of Performance Evaluation
The IVDR has increased the quantity and quality of data required to prove the analytical and clinical performance of IVDs. Manufacturers must submit comprehensive Performance Evaluation Reports demonstrating that their devices provide the intended clinical benefit.
Comparison of Key Changes in MDR and IVDR
The table below summarizes the key changes introduced by the MDR and IVDR, illustrating why the MDR represents a more challenging process for medical device manufacturers:
Feature | MDD (Old System) | MDR (New System) | IVDD (Old System) | IVDR (New System) |
Clinical Evidence | Less detailed, literature was often sufficient. | More Stringent: CER mandatory for every device, higher demand for clinical data. | Self-declaration for most devices. | More Stringent: Significant increase in devices requiring NB oversight due to risk-based classification. |
Notified Body (NB) | Limited role. | Expanded Role: Stricter audits, more devices subject to NB oversight. | Limited role. | Role significantly increased. |
Traceability | Limited. | Mandatory: UDI (Unique Device Identification) and EUDAMED registration. | Limited. | Mandatory: UDI and EUDAMED registration. |
Scope | Did not include some aesthetic devices. | Expanded: Includes Software as a Medical Device (SaMD) and certain aesthetic devices. | Limited. | Expanded: New classification rules and more IVDs brought under regulation. |
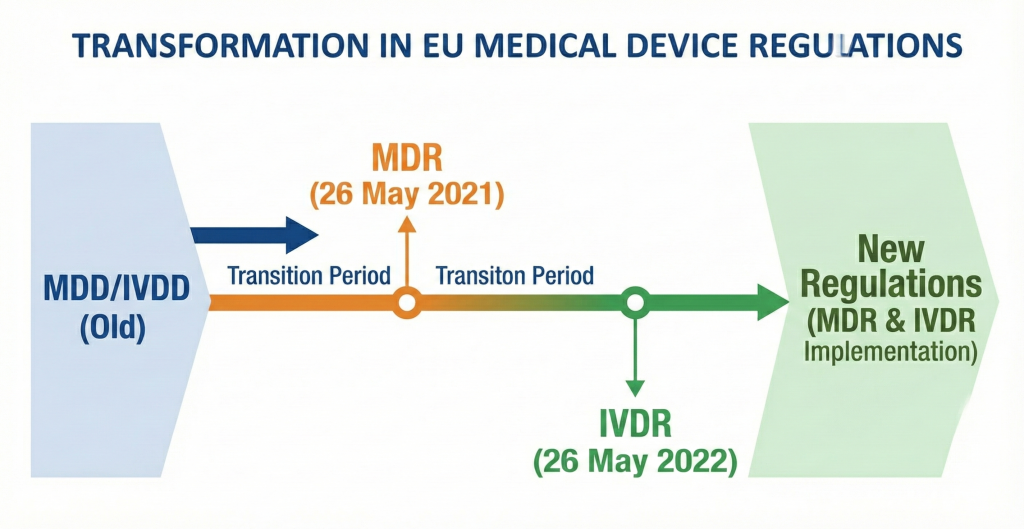
Figure 1: Regulatory Transition Timeline
Conclusion
The MDR and IVDR mark a turning point in the medical device sector. These regulations mandate not only the technical competence of manufacturers but also their proficiency in clinical data collection and post-market surveillance. At MPH CTC, our core mission is to guide you through this complex regulatory environment, ensuring you gather the necessary clinical evidence in the fastest and most effective way.
References
[1] Regulation (EU) 2017/745 of the European Parliament and of the Council of 5 April 2017 on medical devices.
[2] Regulation (EU) 2017/746 of the European Parliament and of the Council of 5 April 2017 on in vitro diagnostic medical devices.
[3] MDCG 2020-1 Guidance on clinical evaluation (MDR).European Commission.