The CE Mark: Passport to the European Market
For medical devices, the CE Mark is a conformity marking that indicates the product complies with the relevant health, safety, and environmental protection standards of the European Union (EU). Under the Medical Device Regulation (MDR), obtaining the CE mark is a mandatory step for a device to be legally sold in the EU market.
While the path to the CE mark varies depending on the device’s risk class (as detailed in Part 3), the fundamental steps apply to all manufacturers.
5 Essential Steps to the CE Mark
Achieving the CE mark requires systematic and disciplined process management.
- Determine the Device’s Scope and Classification
- Clarify whether the device falls under the definition of a medical device and its intended purpose.
- Determine the device’s risk class (Class I, IIa, IIb, III) according to the rules in MDR Annex VIII.
- Establish a Quality Management System (QMS)
- The manufacturer is obliged to establish and maintain a Quality Management System that covers all processes from device design and manufacturing to distribution and post-market surveillance.
- The ISO 13485 standard typically forms the basis for a QMS that meets MDR requirements.
- Prepare the Technical Documentation
- A comprehensive file is created, compiling and organizing all data that proves the device meets the MDR’s general safety and performance requirements (GSPR). (Detailed below.)
- Select the Conformity Assessment Procedure and Notified Body (NB) Process
- Class I (Non-Sterile/Non-Measuring): The manufacturer issues its own Declaration of Conformity based on the Technical Documentation. No NB process is involved.
- Class I (Sterile/Measuring) and Class IIa, IIb, III Devices: The manufacturer selects one of the conformity assessment procedures specified in the relevant MDR annexes and carries out this process with a Notified Body (NB). The NB audits the QMS and reviews the Technical Documentation.
- Issue the Declaration of Conformity and Affix the CE Mark
- After receiving the conformity certificate from the NB (except for Class I), the manufacturer issues the EU Declaration of Conformity, stating that all MDR requirements have been met.
- Finally, the CE Mark is affixed to the device and its packaging.
Technical Documentation: The Heart of MDR Compliance
Technical Documentation (TD) is the most crucial and comprehensive part of MDR compliance. Specified in MDR Annex II and Annex III, this file is a living document that must be kept up-to-date throughout the device’s entire lifecycle.
Section | Content | Importance |
1. Device Description and Specification | Device name, intended purpose, risk class, fundamental design, and operating principle. | Clarifies the device’s identity and scope. |
2. Design and Manufacturing Information | Details of design and manufacturing processes, materials used, production sites. | Demonstrates the applicability of the Quality Management System. |
3. General Safety and Performance Requirements (GSPR) | A list of all GSPRs in MDR Annex I and evidence of how the device meets each requirement. | The core proof of MDR compliance. |
4. Benefit-Risk Analysis and Risk Management | Identification, analysis, evaluation, and control of all risks associated with the device (in compliance with ISO 14971). | Central to the proof of safety. |
5. Verification and Validation Data | Biocompatibility, sterilization, electrical safety, software validation, and most importantly, the Clinical Evaluation Report (CER). | Proves the device’s technical and clinical performance. |
6. Post-Market Surveillance (PMS) Plan | A plan detailing how the device’s safety and performance will be monitored after it is placed on the market. | Evidence of continuous compliance and the lifecycle approach. |
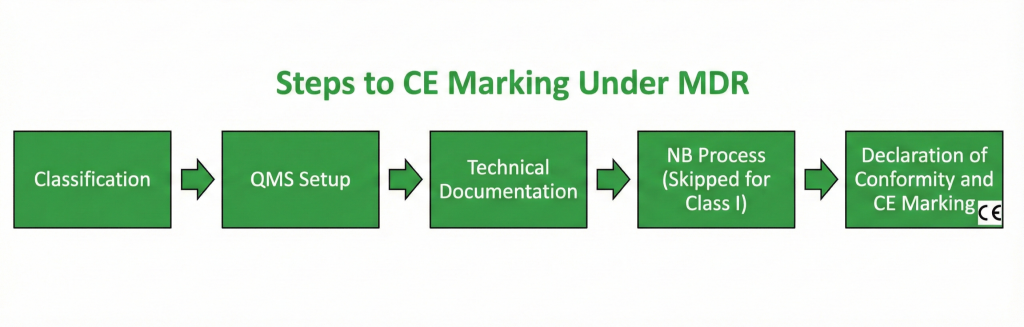
Figure 1: Flowchart of the Path to the CE Mark
MPH CTC’s Role: Accelerating the Process
The most challenging part of the Technical Documentation is the preparation of the Clinical Evaluation Report (CER) and, if necessary, Clinical Investigation data, which proves the device’s clinical performance. At MPH CTC, we step in at this stage to:
- CER Preparation: We align your CER with MDR requirements through comprehensive literature reviews and equivalence analyses.
- Clinical Investigation Management: When new clinical data is required, we plan and execute the investigations in the fastest and most effective way.
By doing so, we ensure that the manufacturer’s Technical Documentation file is complete and audit-ready, thereby accelerating the Notified Body process and shortening the time to achieve the CE mark.
References
[1] Regulation (EU) 2017/745 of the European Parliament and of the Council of 5 April 2017 on medical devices.
[2] MDR Annex II: Technical Documentation.medical-device-regulation.eu.
[3] MDR Annex III: Technical Documentation on Post-Market Surveillance.medical-device-regulation.eu.
[4] ISO 13485:2016 Medical devices — Quality management systems — Requirements for regulatory purposes.