
Clinical Investigation: When is it Unavoidable?
In the previous section (Part 5), we established that the Clinical Evaluation Report (CER) is a systematic analysis of clinical data required to prove the safety and performance of a device. The clinical data needed for the CER is sourced from three main categories: literature, clinical experience, and clinical investigations.
While the MDR encourages manufacturers to primarily use existing data (literature and equivalent device data), in certain situations, collecting new clinical data on their own devices is mandatory. This represents one of the biggest challenges and cost increases introduced by the MDR.
Critical Situations Where Clinical Investigation is Mandatory
The MDR clearly defines the circumstances under which a clinical investigation is mandatory for a device to obtain the CE mark:
Situation | Explanation | Regulatory Impact |
High-Risk Devices (Class III) | Especially for implants and high-risk devices that directly contact the body, when existing literature or equivalent device data is insufficient. | Obligation for the manufacturer to collect new clinical data on their own device. |
Novel Devices | If the device’s technology, intended purpose, or mechanism of action is significantly different from existing devices on the market. | Since a claim of equivalence cannot be established, the device’s safety and performance can only be proven through a new Clinical Investigation (CI). |
Inadequate Equivalence Claim | If the manufacturer cannot prove that their device has the same clinical performance as an equivalent device, or if they do not have full access to the equivalent device’s technical documentation. | The MDR has made the proof of equivalence much more difficult than under the MDD. |
Critical Changes | When significant changes are made to the device’s design or intended purpose that could affect safety or performance. | A new CI may be required to verify the clinical impact of the change. |
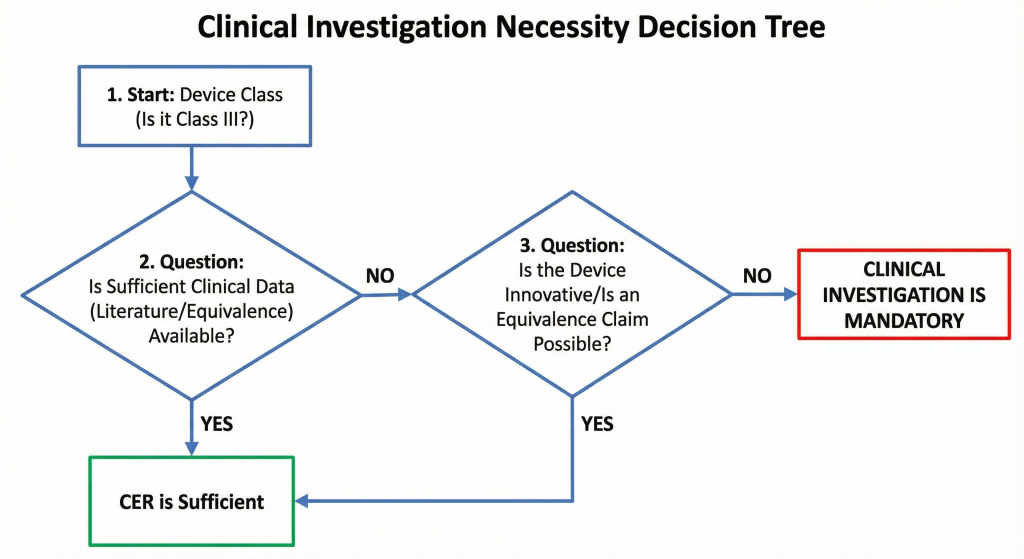
Figure 1. Clinical Investigation Necessity Decision Tree (Flowchart)
Clinical Investigation Procedures: Step-by-Step Roadmap
Clinical investigations must be conducted in accordance with the strict rules set out in MDR Annex XV and the principles of Good Clinical Practice (GCP). The process requires meticulous planning and approval, similar to drug trials.
- Preparation of the Clinical Investigation Plan (CIP)
The CIP is the fundamental document detailing the study’s objective, scientific rationale, methodology, patient selection criteria, monitoring plan, and statistical analysis methods. The CIP ensures the ethical and scientific integrity of the investigation.
- Regulatory and Ethics Approval Submissions
Once the CIP is ready, applications are submitted to the competent authority of the country where the investigation will be conducted (e.g., TİTCK in Turkey) and the local Ethics Committee. These approvals are legal prerequisites for starting the investigation.
- Execution of the Investigation (GCP Implementation)
The investigation is conducted in compliance with the approved CIP and internationally recognized GCP standards. This phase involves patient recruitment, data collection, monitoring, and safety surveillance.
- Data Management and Analysis
All collected data is managed and tracked reliably. Statistical analyses must scientifically prove that the device provides the intended clinical benefit.
- Clinical Investigation Report (CIR)
Upon completion of the investigation, the CIR is prepared, containing all findings, conclusions, and any adverse events. This report is included in the Technical Documentation as the most important and reliable data source for the CER.
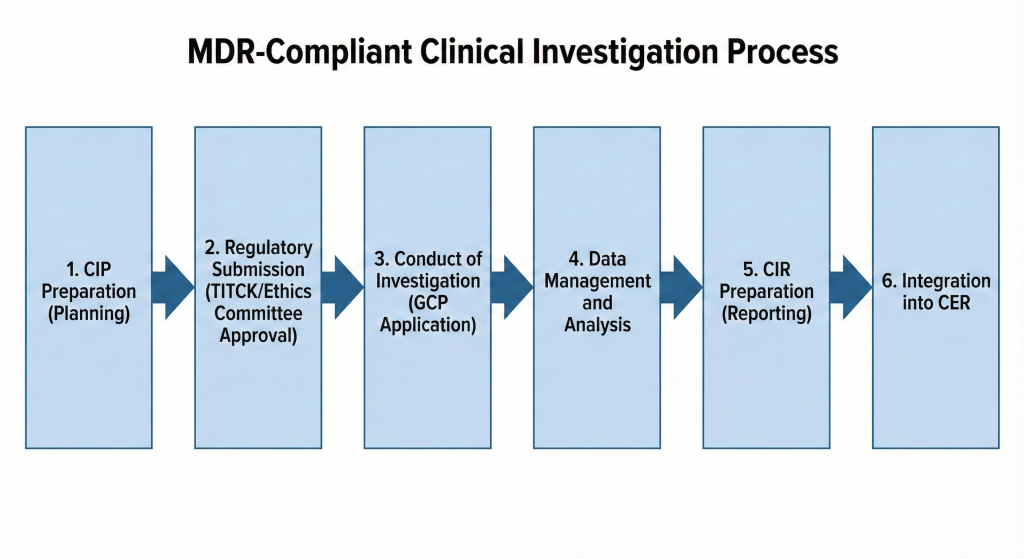
Figure 2. Clinical Research Process Steps (Timeline/Process Diagram)
MPH CTC’s Role in Clinical Investigation: Speed and Compliance
Medical device clinical investigations have different dynamics than drug trials and require specialized expertise. At MPH CTC, we provide critical support to manufacturers in this process:
- Strategic Planning: Designing the CIP to meet MDR requirements and the device’s specific clinical objectives.
- GCP Compliance: Ensuring the investigation is conducted according to international Good Clinical Practice standards, so the collected data is accepted by Notified Bodies without question.
Through this support, manufacturers can complete mandatory clinical investigations in the shortest possible time and with the highest data quality, overcoming a significant hurdle on the path to the CE mark.
References
[1] Regulation (EU) 2017/745 of the European Parliament and of the Council of 5 April 2017 on medical devices, Annex XV.
[2] MDCG 2020-10 Guidance on safety reporting in clinical investigations.European Commission.
[3] ISO 14155:2020 Clinical investigation of medical devices for human subjects — Good clinical practice.